من مرض كرابي هو مرض تخزين وراثي يسبب إزالة الميالين من الجهاز العصبي. سبب هذه الظاهرة هو طفرة صبغية. حتى الآن المرض عضال.
ما هو مرض كرابي؟

© Giovanni Cancemi - stock.adobe.com
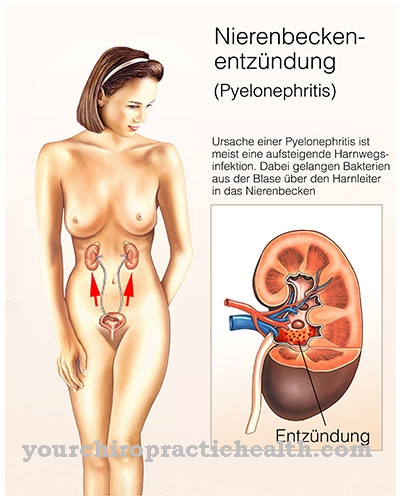
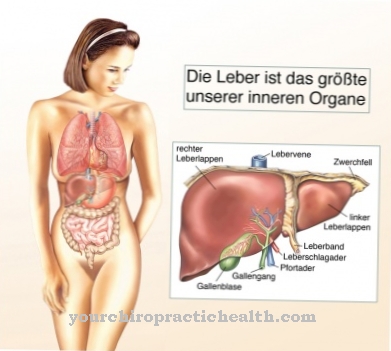
تحت مرض كرابي يفهم الطبيب مرض تخزين نادر من عائلة سيريبروزيد. يسمى المرض أيضا حثل المادة البيضاء للخلايا الكروية معروف. سمي المرض على اسم الطبيب الدنماركي كنود كرابي. كان أول من وصف المرض الوراثي المتنحي بالتفصيل. في مرض كرابي ، تسبب طفرة جينية تراكم مركبات سامة في الجسم. بالإضافة إلى الشكل الطفولي ، هناك شكل خاص من المرض يظهر فقط في مرحلة البلوغ.
التفريق بين مرض كرابي هو مرض جوشر. هذا أيضًا مرض تخزين وراثي يعتمد على طفرة جينية. في كلا المرضين ، يفتقر المصابون إلى الإنزيم. في مرض كرابي ، هو إنزيم β-galactosidase. بدون galactocerebrosidase ، تحدث إزالة الميالين من الجهاز العصبي. من ناحية أخرى ، فإن مرض جوشر يتعلق بإنزيم الجلوكوسيريبروسيداز ، والذي يؤدي نقصه إلى تأثيرات أخرى على الكائن الحي.
الأسباب
سبب مرض كرابي هو خلل في جين GALC. يقع هذا الجين على الكروموسوم 14 ويقع في القسم q3.1. في الترجمة البيولوجية ، يتم ترجمة mRNA إلى بروتينات بناءً على القالب. الجين GALC هو تعليمات بناء لإنزيم galactocerebrosidase أثناء الترجمة. خلل في هذا الجين يسبب أخطاء في الترجمة. في حالة مرض كرابيه ، يحدث حذف ، مما يعني أن الشخص المصاب يفتقر تمامًا إلى إنزيم galactocerebrosidase.
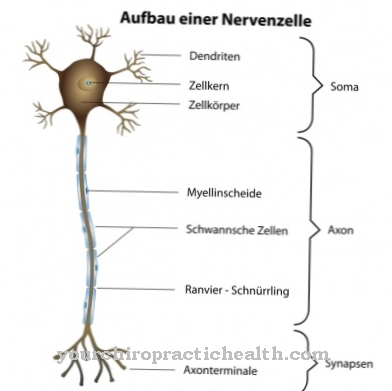
نتيجة لذلك ، تتراكم المواد في الكائن الحي التي تنشأ أثناء استقلاب الميالين. المايلين هو عزل الألياف العصبية في الجهاز العصبي. يقوم إنزيم galactocerebrosidase بتقسيم المنتجات الأيضية للميالين إلى الجالاكتوز والجالاكتوسيريبروسيدات. إذا كان الإنزيم مفقودًا ، فلن يحدث هذا الانهيار بعد الآن. يتراكم Galactocerebroside و psychosin على وجه الخصوص. السيكوسين ضار بالخلايا قليلة التغصن ، مما يضمن الحفاظ على المايلين.
الأعراض والاعتلالات والعلامات
يعاني المرضى المصابون بمرض كرابيه من أعراض مماثلة لمرضى التصلب المتعدد. يرتبط كلا المرضين بزوال الميالين في المسالك العصبية. غالبًا ما يتم ملاحظة إزالة الميالين في البداية كاضطرابات حسية. يقلل انهيار المايلين من سرعة التوصيل العصبي للمريض. اعتمادًا على منطقة الجهاز العصبي المتأثرة حاليًا ، يمكن أن تحدث إعاقات حركية وإدراكية.
يمكن تصور الشلل ، لكن القدرة على الإدراك تكون أيضًا ضعيفة. على سبيل المثال ، قد يحدث العمى في ظل ظروف معينة. الصمم ممكن أيضا. عادة لم يعد من الممكن إثارة ردود أفعال المريض. في داء كرابي الطفولي ، تبدأ الأعراض عادةً بعد بضعة أشهر من العمر. يعاني الرضع المصابون من نوبات صراخ ويسهل غضبهم. غالبًا ما تتمدد الساقين بشكل منشط استجابة للمؤثرات الخارجية. يتوقف التطور ويتم ثني الذراعين باستمرار. يبدأ فرط التنفس والحمى.
التشخيص ومسار المرض
عند تشخيص داء كرابيه ، يجب على الطبيب أن يأخذ في الاعتبار التصلب المتعدد في التشخيص التفريقي. يمكن أن توفر عينة CSF معلومات حول سبب إزالة الميالين. في التصلب المتعدد ، توجد الغلوبولين المناعي في السائل الدماغي النخاعي على الرغم من الحاجز الدموي الدماغي. من ناحية أخرى ، في مرض كرابي ، تزداد البروتينات. يمكن الكشف عن Galactocerebrosidase في الكريات البيض لتأكيد التشخيص.
يمكن أن تساعد مزارع الخلايا الليفية أيضًا في تأكيد التشخيص. الأمر نفسه ينطبق على التحليل الجيني الذي يكشف عن طفرة في الكروموسوم المقابل. إن تشخيص الشكل الطفولي لمرض كرابي غير مواتٍ. يموت الأشخاص المصابون بمتوسط عمر 13 شهرًا. يكون التشخيص أكثر ملاءمة للشكل الخاص في مرحلة البلوغ ، حيث يتقدم المرض بشكل أبطأ في هذه الحالة.
المضاعفات
في معظم الحالات ، يعاني المصابون بمرض كرابيه من اضطرابات مختلفة في الحساسية والشلل. نتيجة لذلك ، فإن الحياة اليومية للمريض مقيدة بشكل كبير ويعتمد المتضررون على مساعدة الآخرين. كما تنخفض جودة حياة المريض بشكل كبير بسبب مرض كرابيه.
ليس من غير المألوف أن يؤدي المرض إلى ضعف إدراكي أو حركي وبالتالي يمكن أن يضعف بشكل كبير نمو الأطفال. علاوة على ذلك ، يمكن أن تحدث المضايقات أو التنمر عند الأطفال ، مما قد يؤدي غالبًا إلى الاكتئاب أو شكاوى نفسية أخرى.
علاوة على ذلك ، يمكن أن يسبب مرض كرابي العمى أو الصمم. يمكن أن يؤدي العمى المفاجئ إلى اكتئاب شديد ، خاصة عند الشباب. غالبًا ما يبكي الأطفال بسبب الأعراض ويعانون أيضًا من الحمى. يمكن للأقارب أو الآباء أيضًا تطوير شكاوى نفسية أو اكتئاب نتيجة المرض.
عادة لا يمكن إجراء العلاج إلا بمساعدة الأدوية. ومع ذلك ، يمكن أيضًا زرع الخلايا الجذعية. لا توجد تعقيدات خاصة. قد يقلل مرض كرابي من متوسط العمر المتوقع للمصابين.
متى يجب أن تذهب إلى الطبيب؟
يمكن التعرف بالفعل على الشكل الكلاسيكي لمرض التخزين مرض كرابي في مرحلة الطفولة المبكرة. إذا علمت الأسرة أن المرض وراثي ، فسوف يراجعون الطبيب بسرعة. خطر إصابة أحد أطفال العائلة بمرض كرابيه مرتفع. ومع ذلك ، فإن الأشقاء يمرضون فقط مع فرصة 1: 4. غالبًا ما يُكتشف داء كرابي أثناء التشخيص قبل الولادة.
نظرًا لأن المرض لا يمكن علاجه ، فإن الإجهاض ممكن إذا تم تحديده طبياً. يكون حثل المادة البيضاء للخلايا الكروية مميتًا دائمًا في الشكل الطفولي. يموت الأطفال عادة قبل بلوغهم سن الواحدة. بصرف النظر عن إعطاء مسكنات الألم لتخفيف الأعراض للأغراض الملطفة ، لم يتبق الكثير ليقوم به الطبيب المعالج. يمكن للطبيب أيضًا أن يصف مستحضرات استرخاء العضلات والمهدئات.
الخيارات العلاجية للأشكال الخاصة من مرض كرابي التي تظهر لاحقًا هي بنفس القدر من السوء. مسار المرض أبطأ ، قد لا تكون الأعراض خطيرة تمامًا. في الشكل المتأخر من مرض كرابيه ، يمكن للأطباء في بعض الأحيان تحقيق تحسينات من خلال زراعة الخلايا الجذعية.
العلاج والعلاج
يعتبر مرض كرابي غير قابل للشفاء. العلاج السببي غير متوفر. على عكس إزالة الميالين من التصلب المتعدد ، لا يمكن تأخير مسار مرض كرابي. بدلاً من إطالة العمر ، ينصب التركيز العلاجي على تحسين نوعية الحياة. لتحقيق هذا الهدف ، غالبًا ما يتم إجراء علاجات دوائية بمضادات التشنج المضادة للتشنج.
يمكن أن يكون لمسكنات الألم والمهدئات آثار أعراض إيجابية. عادة ما يتم رعاية آباء الأطفال المصابين بالعلاج النفسي. في الأشكال المتأخرة من مرض كرابي ، على عكس الشكل الطفولي ، من الممكن حدوث تأخير في مسار المرض. في هذا الصدد ، حققت علاجات الخلايا الجذعية نتائج جيدة. كجزء من هذا العلاج ، يتم نقل خلايا الدم الجذعية من المتبرع إلى المريض.
ومع ذلك ، فإن عملية زرع الخلايا الجذعية الخيفية هي دائمًا عمل محفوف بالمخاطر. على وجه الخصوص ، من الممكن حدوث تفاعلات التهابية وعدوى. مع التقدم في العلاج الجيني ، قد يكون من الممكن في المستقبل علاج المصابين بمرض كرابيه. لكن في الوقت الحالي ، لا يزال هذا السيناريو بعيد المنال.
التوقعات والتوقعات
إن تشخيص مرض كرابي سيئ. يموت العديد من الأطفال في غضون 12 إلى 13 شهرًا من الولادة. يمكن زيادة متوسط العمر المتوقع من خلال زراعة الخلايا الجذعية. علاج المرض الوراثي غير ممكن بعد. لا يمكن علاج الحالة إلا بطريقة تلطيفية. تتطور الأعراض حتى يموت الطفل ، وفي النهاية تحدث حالة غير مستجيبة إلى حد كبير. تحدث الوفاة نتيجة مضاعفات مثل فشل الجهاز التنفسي أو قصور القلب والأوعية الدموية.
يقدم الشكل الطفولي أو الأحداث المتأخر لمرض كرابي تشخيصًا أكثر ملاءمة. يمكن للأطفال الذين يعانون من شكل الأحداث أن يعيشوا عدة سنوات. تظهر الأعراض الأولى في وقت لاحق ، مما يعني أن نمو جهاز المناعة قد اكتمل إلى حد كبير. تفتح الحماية الأفضل للجسم خيارات علاجية إضافية واحتمال حياة خالية من الأعراض نسبيًا.
ومع ذلك ، فإن شكل الأحداث قاتل أيضًا. المرض عبء كبير على الطفل والأقارب. لهذا السبب تتلقى العائلات المتضررة نصائح مفصلة ، يتم خلالها أيضًا مناقشة خيارات التشخيص والعلاج. جمعية ELA Germany e. يقدم V. مزيدًا من التفاصيل حول تشخيص مرض كرابي.
منع
لا يمكن منع طفرات الكروموسومات مثل مرض كرابي مباشرة. ومع ذلك ، يمكن للعائلات التي تخطط لأطفالها استخدام التحليل المتسلسل للحمض النووي الخاص بهم لتقييم مخاطر نقل الأمراض الوراثية. إذا كان الطفل مصابًا بمرض كرابي ، فهناك احتمال بنسبة 25 في المائة للأطفال الآخرين.
الرعاية اللاحقة
مرض كرابي غير قابل للشفاء حاليًا باعتباره حالة وراثية ، وبالتالي لا توجد متابعة طبية بالمعنى الضيق. الدورة تنتهي دائما بموت. إذا حدثت الحالة في مرحلة الطفولة أو بعد الولادة ، يجب أن يكون الوالدان على دراية جيدة بمرض كرابي والعلاج. عندها فقط يهدف علاج الأعراض في المقام الأول إلى السيطرة على التهيج والشلل التشنجي. في المراحل اللاحقة من المرض ، تعتبر الإجراءات الملطفة هي الأكثر أهمية.
الأعراض هي نفسها عند الأشخاص الذين يصابون بالمرض لاحقًا. ومع ذلك ، فإن العملية أبطأ ، مما يمنح الأقارب مزيدًا من الوقت للاستعداد وقضاء الوقت مع الشخص المصاب بمرض كرابيه. يتم تقديم رعاية المتابعة للأقارب في شكل رعاية نفسية ، إذا كان ذلك مطلوبًا. يمكن أن يوفر الاختبار الجيني أيضًا معلومات حول مدى خطورة تعرض الأبناء لمزيد من مرض كرابي.
يمكن تصور زرع الخلايا الجذعية. ومع ذلك ، لا يمكن تحقيق علاج من خلال هذا أيضًا. في حالة محاولة زرع الخلايا الجذعية ، يلزم أيضًا اتخاذ تدابير الرعاية اللاحقة اللازمة. وهذا يعني ، على سبيل المثال ، حماية خاصة للمتلقي ضد الجراثيم ، لأن جهاز المناعة لديه يجب أن يضعف من أجل العلاج.
يمكنك أن تفعل ذلك بنفسك
لم يتم الشفاء من مرض كرابي بعد. لذلك يحتاج آباء الأطفال المصابين في كثير من الأحيان إلى دعم علاجي. يمكن دعم العلاج بالعقاقير من خلال تدابير مختلفة. بادئ ذي بدء ، يجب مراقبة الطفل على مدار الساعة حتى يمكن استخدام الدواء اللازم بسرعة في حالة حدوث تقلصات. بالإضافة إلى مسكنات الألم ، يمكن أن توفر التمارين والتشتيت أيضًا الراحة.
يجب دائمًا علاج الإعاقات الحركية والإدراكية النموذجية في عيادة متخصصة. يجب على الآباء التحدث إلى الطبيب المسؤول عن المزيد من خيارات العلاج ، حيث تتقدم الأبحاث حول المرض الوراثي النادر بسرعة ويتم العثور باستمرار على طرق علاج جديدة. يمكن علاج التصلب المتعدد الفعلي بالعلاج الطبيعي والتدليك وتمارين الاسترخاء المختلفة. هذا يخفف بشكل فعال الأعراض النموذجية مثل الرعاش أو الدوخة. يمكن أن تساعد تمارين قاع الحوض المنتظمة في حل مشاكل الفاعلية.
يمكن أيضًا تحسين نوعية حياة الطفل والوالدين من خلال الإرشاد النفسي الشامل. خاصة بعد فترة طويلة من المرض ، من الضروري مناقشة الوضع المعيشي مع أخصائي وتطوير أساليب علاجية جديدة. في الوقت نفسه ، يجب تنفيذ المهام التنظيمية مثل التسجيل في رياض الأطفال الخاصة وشراء الأدوات المساعدة.
.jpg)
.jpg)

.jpg)