من مرض نيمان بيك هو أيضا مرض نيمان بيك معروف. المرض الوراثي هو أحد أمراض التخزين الليزوزومي.
ما هو مرض نيمان بيك؟

© ktsdesign - stock.adobe.com
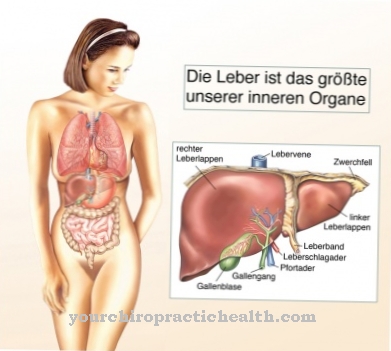
من مرض نيمان بيك هو مرض من فصيلة الشحميات السفينغولية. هذه هي أمراض التمثيل الغذائي التي تظهر في الغالب في الجهاز العصبي المركزي. داخل الجسيمات الشحمية السفينغولية ، ينتمي المرض إلى أمراض التخزين الليزوزومية. تتميز هذه بخلل في الجسيمات الحالة.
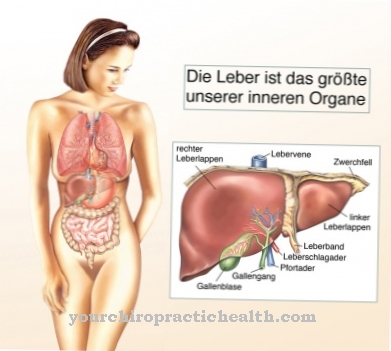
في البلدان الناطقة باللغة الإنجليزية ، مصطلح أمراض التخزين الليزوزومية (LSDs) تستخدم. في مرض Niemann-Pick ، يتم ترسيب السفينجوميلين في الكبد ونخاع العظام والطحال والدماغ. سمي المرض باسم مكتشفيه ألبرت نيمان ولودفيج بيك. تم وصفه لأول مرة في عام 1914. نادرًا ما يحدث مرض Niemann-Pick.
سيصاب حوالي طفل واحد من بين كل 8000 ولادة بمرض التخزين الليزوزومي. لكن هذا لا يشمل ذلك فقط مرض نيمان بيك، ولكن أيضًا أمراض مثل متلازمة هنتر أو متلازمة سانفيليبو.
الأسباب
يُورث مرض Niemann-Pick كصفة جسمية متنحية. في الوراثة المتنحية الجسدية ، يكون الأليل المعيب على كروموسوم متماثل أو صبغي جسدي. تمرض فقط ناقلات متماثلة اللواقح من السمة. هذا يعني أن المادة الجينية للخلية يجب أن تحتوي على نسختين متطابقتين من الجين المعيب على كلا الكروموسومين حتى ينتشر المرض.
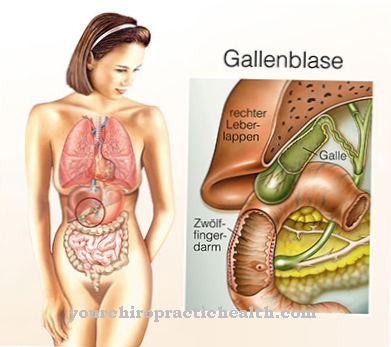
تعتمد متلازمة Niemann-Pick على عيب إنزيم وراثي. يتأثر إنزيم السفينغوميليناز. إن sphingomyelinase هو المسؤول عن انقسام sphingomyelin. يؤدي عيب الإنزيم إلى زيادة تخزين السفينجوميلين في الجسيمات الحالة في الطحال ونخاع العظام والدماغ والكبد. الليزوزومات هي عضيات خلوية تحتوي على إنزيمات هضمية.
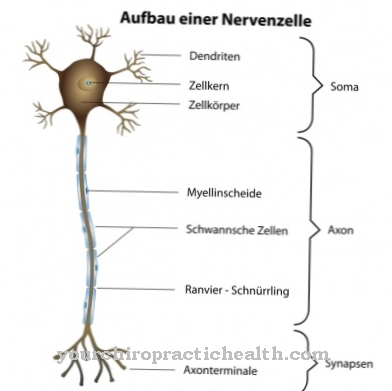
يهضمون المواد الغريبة مثل مسببات الأمراض أو بقايا الخلايا. كما أنها تلعب دورًا مهمًا في موت الخلايا المبرمج (موت الخلايا المبرمج). في التجارب على الحيوانات ، يمكن إثبات أن التعبير عن العامل التنظيمي لجين المايلين (MRF) قد انخفض بشكل كبير من خلال الطفرة في الجين NPC-1. بروتين MRF هو ما يسمى عامل النسخ. في الترميز الجيني ، يلعب دورًا في تكوين وحماية أغلفة المايلين.
تغطي أغلفة المايلين الألياف العصبية وتضمن تمرير المنبهات بسرعة. من المفترض أن العيوب العصبية التي تحدث في مرض Niemann-Pick تستند إلى تمايز غير صحيح للخلايا قليلة التغصن. تنتمي هذه الخلايا إلى الخلايا الدبقية. تغطي عمليات الخلايا الخاصة بهم عمليات الخلايا للألياف العصبية كأغماد المايلين. وبالتالي ، فإن التمايز الخاطئ للخلايا قليلة التغصن يؤدي إلى نقص أو نقص النخاع.
في حالة مرض Niemann-Pick من النوع C ، يتأثر أيض الكوليسترول أيضًا. بالإضافة إلى السفينجوميلين ، يتراكم الكوليسترول ومنتجات التمثيل الغذائي الأخرى أيضًا في خلايا الجسم.
الأعراض والاعتلالات والعلامات
يمكن تقسيم مرض Niemann-Pick إلى ثلاثة أشكال:
- يُعرف النوع IA أيضًا باسم شكل الاعتلال العصبي عند الأطفال. يبدأ المرض في عمر ثلاثة أشهر ويتجلى على أنه ضعف في الشرب واضطرابات في النمو للأنسجة والأعضاء الفردية.
يتمثل العرض الرئيسي في تورم الكبد (تضخم الكبد). يمكن أن يحدث هذا أيضًا مع تورم الطحال (تضخم الطحال). بالإضافة إلى ذلك ، يمكن الشعور بالعقد الليمفاوية ويحدث تلون بني للجلد. يبدأ التدهور العصبي في السنة الثانية من العمر. يصاب الأطفال الصغار بالصمم والعمى ويفقدون الاتصال الاجتماعي.
التكهن ضعيف ، مما يعني أن جميع الأطفال المصابين بمرض Niemann-Pick من النوع IA سيموتون في غضون عامين. هذا الشكل هو البديل الأكثر شيوعًا للمرض.
- يُعرف TYPE IS أيضًا بالشكل الحشوي المزمن. وهي دورة معتدلة مع تورم الكبد وتسلل الرئة. لا يوجد تدخل للجهاز العصبي المركزي. متوسط العمر المتوقع للمرضى مقيد قليلاً فقط.
- في النوع C من مرض Niemann-Pick ، يحدث اليرقان الوليدي. يتحول الجلد والصلبة عند الأطفال حديثي الولادة المصابين إلى اللون الأصفر بفعل رواسب البيليروبين الصبغية. يعد الشلل فوق النووي أيضًا نموذجيًا لهذا النوع من المرض. وهذا يؤدي إلى شلل تدريجي في عضلات العين مع ازدواج الرؤية أو اضطرابات في التوازن.
يمكن أيضًا ملاحظة رنح المخيخ مع ضعف تنسيق الحركة. أثناء المرض ، غالبًا ما يصاب المرضى باضطرابات في البلع. هذا يمكن أن يسبب الالتهاب الرئوي التنفسي. بداية المرض في النوع C متغيرة للغاية. يمكن أن تظهر الأعراض الأولى عند الرضع أو الأطفال أو حتى في مرحلة المراهقة أو البلوغ.
التشخيص ومسار المرض
إذا كان خطر المرض معروفًا ، فمن الممكن تشخيص ما قبل الولادة. في حالة الاشتباه في مرض نيمان-بيك ، يتم أخذ خلايا الدم البيضاء من نخاع العظم. تبدو هذه مفرغة. هذا يعني أن الكريات البيض بها تجاويف. هناك أيضًا خلايا رغوية مفرغة.
تُعرف هذه الظاهرة باسم "كثرة المنسجات الأزرق البحري". يمكن الكشف عن نقص نشاط إنزيم sphingomyelinase في مزارع الكريات البيض والخلايا الليفية. يظهر كل طفل مصاب بمرض Niemann-Pick علامة البقعة الصفراء الحمراء أثناء قاع العين.
المضاعفات
اعتمادًا على النوع ، يرتبط مرض Niemann-Pick بعدد من المضاعفات. مع TYPE IS ، يمكن أن يحدث تورم الكبد وتسلل الرئة ، أي تراكمات الأجسام الغريبة في الرئتين. يتم تقييد متوسط العمر المتوقع للمتضررين بشكل طفيف وتتأثر جودة الحياة في بعض الأحيان بشدة. مع النوع C ، يمكن أن تظهر الأعراض الأولى في مرحلة الطفولة.
يمكن أن يؤدي هذا إلى اضطرابات نمو شديدة ، والتي غالبًا ما ترتبط بالرنح المخيخي مع اضطرابات تنسيق الحركة. أثناء المرض تحدث أحيانًا اضطرابات في البلع ، مما يؤدي إلى التهاب رئوي تنفسي ومضاعفات أخرى. تظهر على المصابين في بعض الأحيان أعراض ضيق في التنفس ، والذي يرتبط بالسعال مع البلغم ، وزيادة درجة حرارة الجسم ، وتغير لون الجلد والأغشية المخاطية إلى اللون الأزرق.
في المقابل ، فإن مثل هذا الزرقة محفوف بمضاعفات خطيرة. في النوع IA ، هناك ضعف مبكر في الشرب واضطرابات في نمو الأعضاء والأنسجة. عادة ما يرتبط تورم الكبد بتورم الطحال ، مما يسبب ضعفًا جسديًا شديدًا لدى المصابين.
تحدث العدوى بشكل متكرر ، ويلتهب الجهاز الهضمي وتنخفض وظائف الجسم بسرعة. عادة ما يصاب الأطفال الصغار بالصمم والعمى في غضون عامين قبل أن يموتوا أخيرًا بسبب المضاعفات الشديدة لمرض نيمان-بيك.
متى يجب أن تذهب إلى الطبيب؟
مرض Niemann-Pick هو مرض وراثي يأخذ مسارًا تدريجيًا. يجب على الآباء الذين يجدون أن طفلهم يعاني من اليرقان المتكرر وانزعاج العضلات الاتصال بطبيب الأطفال. إذا كان هناك تأخيرات في النمو الحركي أو اضطرابات سلوكية نفسية ، فإن الاشتباه في وجود مرض خطير يحتاج إلى التشخيص والعلاج يكون واضحًا.
يجب على الآباء أو الأوصياء زيارة مركز متخصص للأمراض الأيضية النادرة. يحتاج الأطفال المصابون بمتلازمة نيمان-بيك إلى علاج طبي مستمر بسبب زيادة المشكلات الجسدية والعقلية.
يجب إبلاغ الطبيب المسؤول بالأعراض غير العادية أو الزيادة المفاجئة في الأعراض النموذجية. وينطبق الشيء نفسه إذا لم يعد الطفل قادرًا على تحمل الدواء الموصوف أو أظهر انحرافات أخرى عن السلوك الطبيعي. يمكن أن يقوم طبيبك بإجراء العلاجات الروتينية مثل إيقاف الأدوية والفحوصات الجسدية.
يجب أن يعالج معظم الأشخاص المصابين بمرض Niemann-Pick من قبل متخصصين في أمراض التمثيل الغذائي. يتم علاج الأعراض الفردية من قبل أطباء الأعصاب وجراحي العظام وأخصائيي النطق. بالإضافة إلى ذلك ، يشارك أخصائيو العلاج الطبيعي والمعالجون المهنيون في العلاج. يمكن أيضًا استدعاء المعالج للشكاوى النفسية مثل الاكتئاب أو الأوهام. نظرًا للعدد الكبير من الأعراض المحتملة ، يجب عادةً علاج مرض Niemann-Pick من قبل فريق من الأطباء.
العلاج والعلاج
العلاج السببي غير معروف حاليًا. ومع ذلك ، هناك أدلة على أن السيكلودكسترين الخاص يمكن أن يخفف من أعراض المرض. الدكسترين الحلقي عبارة عن سكريات قليلة السكاريد الحلقية تستخدم غالبًا كمذيبات في إنتاج الأدوية. يُعالج مرض Niemann-Pick من النوع C بالهجين.
Miglustat هو دواء معتمد فقط في الاتحاد الأوروبي لعلاج مرض Niemann-Pick ولعلاج مرض Gaucher من النوع 1. الدواء هو iminosugar ومشتق n-butyl من المورانولين.
يمكنك العثور على أدويتك هنا
- أدوية للألمالتوقعات والتوقعات
إن تشخيص مرض Niemann-Pick ضعيف. المرض هو خلل جيني. يحظر التشريع الحالي على العلماء التدخل في الجينات البشرية أو تعديلها. على الرغم من أنه يمكن تشخيص المرض قبل الولادة ، فلا يوجد علاج ممكن بناءً على المتطلبات القانونية.
حتى يومنا هذا ، يركز الأطباء والمهنيون الطبيون على تطوير رعاية طبية كافية بعد ولادة الشخص. يتكون العلاج حاليًا من بدء العلاج الدوائي من أجل دعم عملية التمثيل الغذائي للمريض على أفضل وجه ممكن. ونتيجة لذلك ، فإن التحسينات ممكنة بالفعل في عملية تطوير المريض ، مما يساهم في تحسين الوضع العام.
بدون علاج ، تنخفض جودة حياة الشخص المصاب بشكل كبير. بالإضافة إلى ذلك ، يمكن أن تتطور الحالات التي تهدد الحياة ، حيث يصاحب المرض تورم في الأعضاء الداخلية وضيق في التنفس. يزداد خطر حدوث حالة طارئة بشكل كبير دون علاج. لذلك يُشار إلى العلاج طويل الأمد بغض النظر عن شدة الأعراض الفردية. يحتاج المرضى إلى رعاية ودعم يومي في التعامل مع الحياة اليومية. اعتمادًا على نوع المرض الموجود ، إذا تقدم المرض بشكل سيئ ، فقد يموت المريض قبل الأوان خلال السنوات القليلة الأولى من الحياة.
منع
يُورث مرض Niemann-Pick كصفة وراثية متنحية. لا يوجد حاليا أي وقاية فعالة.
الرعاية اللاحقة
في معظم الحالات ، يكون لدى الشخص المصاب عدد قليل ومحدود فقط من إجراءات المتابعة المتاحة لمرض Niemann-Pick. لهذا السبب يجب على المريض استشارة الطبيب عند ظهور العلامات والأعراض الأولى حتى لا تكون هناك مضاعفات أو شكاوى أخرى. كلما تم الاتصال بالطبيب مبكرًا ، كان المسار المستقبلي للمرض أفضل ، بحيث يجب استشارة الطبيب بمجرد ظهور الأعراض أو العلامات الأولى.
إذا كان المريض يرغب في إنجاب الأطفال ، فيجب إجراء الاختبارات الجينية والاستشارة من أجل منع تكرار مرض Niemann-Pick. يعتمد معظم المرضى عادة على تناول الأدوية المختلفة.
يجب أن ينتبه الشخص المصاب دائمًا إلى الجرعة الصحيحة وأيضًا إلى تناوله بانتظام من أجل تخفيف الأعراض بشكل دائم. إذا كان أي شيء غير واضح أو إذا كان لديك أي أسئلة ، فيجب دائمًا استشارة الطبيب أولاً. وبالمثل ، يعتمد العديد من المرضى على مساعدة ودعم أسرهم في حياتهم اليومية. وفوق كل شيء ، يمكن التخفيف من حدة الاكتئاب والشكاوى النفسية الأخرى.
يمكنك أن تفعل ذلك بنفسك
إمكانيات المساعدة الذاتية محدودة للغاية مع مرض Niemann-Pick. النوع IA على وجه الخصوص لا يوفر فرصًا كافية لتحسين الوضع. متوسط العمر المتوقع للطفل المريض منخفض للغاية على الرغم من كل الجهود.
لذلك يجب أن يكون التركيز في الحياة اليومية على جعل الوقت معًا ممتعًا قدر الإمكان. الاستمتاع بوقت الفراغ مهم لبناء التقارب والتضامن والاستقرار. يمثل المرض تحديًا هائلاً لكل من المرضى والأقارب. بناء القدرات العقلية مهم بشكل خاص عند التعامل مع المحن. لهذا السبب ، يعد الدعم النفسي ضروريًا لجميع المعنيين.
بالنسبة للكثيرين ، من المفيد أيضًا إذا كان هناك إمكانية للتبادل مع الأشخاص المتضررين الآخرين. لذلك قد يكون من المفيد الاتصال بمجموعات المساعدة الذاتية القائمة. في المناقشات المشتركة ، يتم التبادل على أساس التفاهم المتبادل. يمكن أن يساعد الاتصال في المعالجة. كما أنه يعطي نصائح للتأقلم بشكل جيد.
تساعد التقنيات العقلية وتمارين الاسترخاء على تقليل الضغوطات. نظرًا لظهور حالات الإجهاد المفرط وبالتالي المشكلات الخضرية ، يمكن أن تساعد وحدات التدريب في تقليل التوتر. وبالتالي يجب تحسين التعامل مع الوضع العام.

.jpg)