في ال نقص α-N-acetylgalactosaminidase إنه مرض تخزين ليزوزومي نادر الحدوث. وهي مقسمة إلى حدث وشكل بالغ.
ما هو نقص a-N-Acetylgalactosaminidase؟

نقص a-N-acetylgalactosaminidase أو نقص Alpha-N-acetylgalactosaminidase يسمى أيضا في الطب مرض شندلر, مرض شندلر أو مرض كانزاكي المحددة. والمقصود مرض خزن جسيمي شديد الندرة ، وراثة وراثة جسمية متنحية. يفرق الأطباء بين شكل الأحداث ، الذي يحدث عند الأطفال والمراهقين ، وشكل البالغين الذي يعاني منه البالغون.
يسمى الشكل الأحداث من نقص alpha-N-acetylgalactosaminidase بمرض شندلر ، بينما يسمى الشكل البالغ بمرض كانزاكي. ولكن هناك أيضًا تقسيمات فرعية لمرض شندلر من النوع الأول للشكل الشاب ومرض شندلر من النوع الثاني لشكل البالغين. مزيج من كلا الشكلين ، المعروف باسم مرض شندلر من النوع الثالث ، ممكن أيضًا.
جميع أشكال المرض الثلاثة هي السكريات قليلة السكاريد (البروتينات السكرية) ، والتي تشمل داء الفوكوزيدات ومرض تخزين حمض السياليك. يعود اسم مرض شندلر أو مرض شندلر الناجم عن نقص alpha-N-acetylgalactosaminidase إلى عالم الوراثة الألماني ديتليف شندلر.
كان أول من وصف المرض في عام 1988. بعد عام ، وصف الطبيب الياباني ت. كانزاكي شكل البالغين. اكتشف في عام 1991 أن نقص alpha-N-acetylgalactosaminidase هو سبب المرض.
الأسباب
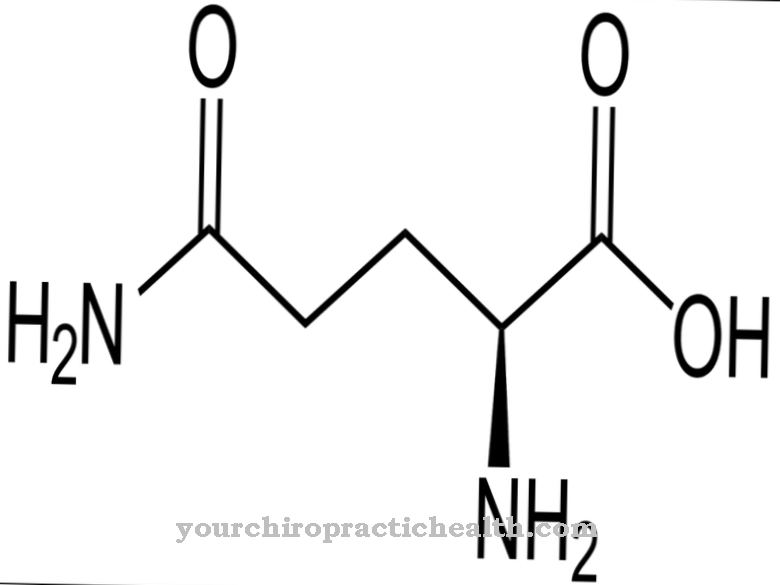
يحدث نقص α-N-acetylgalactosaminidase بسبب انخفاض نشاط إنزيم alpha-N-acetylgalactosaminidase. الطفرات الخاطئة أو غير المنطقية على جين NAGA ، والتي ترمز لـ alpha-N-acetylgalactosaminidase ، هي المسؤولة عن عيب الإنزيم.
يقع جين NAGA في موضع الجين الكروموسوم 22 q11. يقوم إنزيم alpha-N-acetylgalactosaminidase بمهمة تحفيز انقسام N-acetylgalactosamine من مختلف الجليكوليبيدات والبروتينات السكرية. يتسبب الخلل الجيني في انخفاض نشاط الإنزيم ، بحيث تتراكم المواد التي لا يتم استقلابه في خلايا الجسم.
في معظم الحالات ، تكون هذه البروتيوغليكان ، والجليكوسفينجوليبيدات ، والبروتينات السكرية المرتبطة بـ N أو O. يؤدي تراكم هذه المواد إلى تلف الخلية والأعضاء المصابة. يعد نقص Alpha-N-acetylgalactosaminidase نادرًا جدًا بحيث لا توجد معلومات دقيقة حول تردده.
حتى الآن ، من المعروف أن اثني عشر مريضًا فقط من ثماني عائلات أصيبوا بمرض شندلر في العالم. لا تحدث شكاوى عصبية عند كل مريض. يشتبه بعض الأطباء في تأثير عوامل إضافية لحدوث الأعراض العصبية. ومع ذلك ، لا يوجد حتى الآن أي دليل يدعم هذا الافتراض.
يمكنك العثور على أدويتك هنا
- أدوية للألمالأعراض والاعتلالات والعلامات
تعتمد أعراض نقص alpha-N-acetylgalactosaminidase على الشكل المعين للمرض. في سياق مرض شندلر من النوع الأول ، يحدث نقص توتر العضلات التدريجي في السنة الأولى من العمر. بالإضافة إلى ذلك ، يعاني الأطفال المصابون من تراجع واضح في الحركة النفسية.
كما تم تسجيل الاضطرابات في تسلسل الحركة والشلل الرباعي التشنجي والنوبات الدماغية رمعية عضلية كأعراض. هناك أيضًا خطر إصابة الطفل بالعمى. إذا كان هناك شكل بالغ من نقص alpha-N-acetylgalactosaminidase ، أي مرض شندلر من النوع 2 ، تظهر أعراض مشابهة لمرض فابري.
يعاني المرضى من أورام وعائية ، وهي تغيرات جلدية حميدة. كثير من المرضى متخلفون عقليا قليلا. الشكل الإكلينيكي الوسيط هو مرض شندلر من النوع الثالث ، ويعاني المصابون من خلل عصبي ونوبات دماغية وإعاقات عقلية.
من الممكن أيضًا وجود أشكال أقل حدة ، والتي ترتبط بشكاوى نفسية وعصبية خفيفة ، وتأخر تطور اللغة أو أعراض مشابهة لمرض التوحد.
التشخيص والدورة
يمكن تشخيص نقص alpha-N-acetylgalactosaminidase من خلال الكشف عن نشاط NAGA المنخفض. لهذا الغرض ، يتم إجراء اختبارات الإنزيم في بلازما الدم أو الكريات البيض أو الخلايا الليفية المستزرعة أو الأرومات اللمفاوية. يتيح الكشف الكروماتوغرافي للسكريات قليلة التعدد في البول أيضًا تشخيص نقص الإنزيم.
يمكن أيضًا إجراء تحليل الحمض النووي ، لكن هذا ليس ضروريًا في معظم الحالات. أثناء الحمل ، يمكن أيضًا إجراء التشخيص السابق للولادة لنقص الإنزيم باستخدام تحليل الطفرات في جين NAGA بعد أخذ عينة من الزغابات المشيمية أو بزل السلى. ومع ذلك ، لا يمكن تحديد الشكل المناسب للمرض.
من المهم أيضًا التشخيص التفريقي لمتلازمة فابري ، والتنكس العصبي المرتبط بالبانثوثينات كيناز وضمور المحور العصبي الطفلي ، والتي تحتوي أيضًا على عيوب إنزيمية. يعتمد مسار نقص alpha-N-acetylgalactosaminidase على شكل المرض المعني.
يعتبر مرض شندلر من النوع الأول غير مواتٍ ، في حين أن تشخيص النوعين الآخرين يكون أكثر ملاءمة. ومع ذلك ، فإن عدد المرضى ضئيل للغاية بحيث لا يمكن الإدلاء ببيانات ذات مغزى حول متوسط العمر المتوقع.
المضاعفات
تعتمد أعراض ومضاعفات نقص A-N-acetylgalactosaminidase بشكل كبير على شكله.ومع ذلك ، تحدث اضطرابات الحركة في معظم الحالات ، بحيث يتم تقييد المصابين نسبيًا في حياتهم اليومية. تحدث أيضًا تقلصات مرتبطة بألم شديد.
يعاني الأطفال على وجه الخصوص من اضطرابات النمو الشديدة ، والتي يمكن أن تؤدي إلى التشنج لدى الطفل. في بعض الحالات ، يؤدي نقص A-N-acetylgalactosaminidase إلى العمى التام لدى المريض. يؤدي تراجع الذكاء إلى التخلف وبالتالي إلى الإعاقة.
هذه عادة ما تقيد الحياة اليومية للمريض بشكل كبير ولها تأثير سلبي على نوعية الحياة. غالبًا ما يعتمد المصابون على مساعدة الآخرين. يمكن أن يؤدي التخلف أيضًا إلى اضطراب في العثور على الكلمات أو اضطراب لغوي. لا يمكن معالجة نقص A-N-acetylgalactosaminidase سببيًا.
لهذا السبب ، يجب على الشخص المصاب تناول المضادات الحيوية. يجب أيضًا تجنب بعض المضاعفات بشكل مباشر. الالتهاب الرئوي ، على سبيل المثال ، هو واحد من هؤلاء. علاوة على ذلك ، يمكن أن يعتمد المريض أيضًا على التغذية الاصطناعية.
متى يجب أن تذهب إلى الطبيب؟
يجب أن يعالج الطبيب بالتأكيد نقص A-N-acetylgalactosaminidase. هذا النقص لا يزول من تلقاء نفسه ولا يوجد شفاء تلقائي لهذا المرض. على أي حال ، يجب استشارة الطبيب إذا لاحظ الوالدان انخفاضًا في القدرات الحركية والعقلية لدى طفلهما أو طفلهما. يمكن أن تشير الشكاوى المتعلقة بالحركات أو العمليات البسيطة أيضًا إلى نقص A-N-acetylgalactosaminidase ويجب فحصها من قبل الطبيب.
علاوة على ذلك ، يعتبر التشنج المتنوع أيضًا علامة على المرض. ليس من غير المألوف أن يعاني المرضى من إعاقات ذهنية وأعطال أخرى. في حالة وجود هذه الشكاوى ، يكون العلاج ضروريًا بالتأكيد. في حالة عدم وجود علاج ، يمكن أن يؤدي ذلك إلى شكاوى ومضاعفات كبيرة في مرحلة البلوغ وبالتالي تقليل جودة حياة الشخص المصاب بشكل كبير.
يمكن أن يكون التأخر الشديد في تطور الكلام أيضًا علامة على نقص A-N-acetylgalactosaminidase وبالتالي يجب التحقيق فيه. كقاعدة عامة ، يمكن استشارة طبيب عام لتحديد نقص A-N-acetylgalactosaminidase. ثم يتم إجراء المعالجة الإضافية للشكاوى الفردية من قبل أخصائي.
الأطباء والمعالجين في منطقتك
العلاج والعلاج
العلاج السببي لنقص alpha-N-acetylgalactosaminidase غير ممكن. لذلك ، يقتصر النهج الطبي على علاج الأعراض. ويشمل ذلك اتباع نظام غذائي معقول وتناول السوائل ، والوقاية من الأمراض المعدية ، والتي يمكن القيام بها أيضًا عن طريق إعطاء المضادات الحيوية ، واحتواء النوبات الدماغية عن طريق إعطاء الأدوية المضادة للصرع.
تشمل خطوات العلاج الإضافية تدابير العلاج الطبيعي للوقاية من الالتهاب الرئوي والتقلصات ، ولتقليل الألم أو التشنج بمساعدة الأدوية. علاوة على ذلك ، يمكن أن تتم الوقاية من الشفط من خلال التغذية الاصطناعية.
أظهرت الدراسات الحديثة أن نقص α-N-acetylgalactosaminidase هو اضطراب في طي البروتين ، لذلك تتم مناقشة العلاج ببدائل الإنزيم كإجراء علاجي معقول. العلاج الجيني هو نهج علاجي آخر يمكن تصوره لعلاج اضطرابات التخزين الليزوزومية. نظرًا لأن مرض شندلر موروث بطريقة وراثية متنحية ، يوصى بالاستشارة الوراثية للعائلات المصابة.
التوقعات والتوقعات
يمكن أن يؤدي نقص A-N-acetylgalactosaminidase إلى أعراض مختلفة. ومع ذلك ، في معظم الحالات ، يؤدي النقص إلى تأخر النمو الحركي النفسي. في مرحلة البلوغ ، غالبًا ما يعتمد المريض على مساعدة الأشخاص الآخرين ولا يمكنه التعامل مع الحياة اليومية بمفرده. تحدث اضطرابات الحركة أيضًا. يمكن ملاحظة ذلك من خلال عرج أو اضطرابات في التنسيق.
في أسوأ الحالات ، يمكن أن يصاب المريض بالعمى أو يعاني من اضطرابات بصرية شديدة بسبب نقص A-N-acetylgalactosaminidase. ليس من غير المألوف أن تحدث الإعاقات الذهنية التي تقلل إلى حد كبير من نوعية حياة الشخص المعني. تحدث أيضًا اضطرابات الكلام واضطرابات البحث عن الكلمات ، مما قد يؤثر على الحياة. يتأثر الأطفال على وجه الخصوص بالتنمر والمضايقة بسبب أعراض نقص A-N-acetylgalactosaminidase.
العلاج السببي لنقص A-N-acetylgalactosaminidase غير ممكن ، لذا فإن العلاج يهدف في المقام الأول إلى تقليل الأعراض. يمكن استخدام تدابير علاجية مختلفة للحد من اضطرابات النمو. ومع ذلك ، في كثير من الحالات ، يعتمد المريض على المساعدة الخارجية في الحياة اليومية. متوسط العمر المتوقع لا يتأثر بالنقص.
يمكنك العثور على أدويتك هنا
- أدوية للألممنع
لا توجد تدابير وقائية معروفة ضد نقص alpha-N-acetylgalactosaminidase. مرض شندلر هو أحد الأمراض النادرة للغاية.
الرعاية اللاحقة
نظرًا لأن نقص A-N-acetylgalactosaminidase هو مرض خلقي لا يمكن علاجه سببيًا ولكن عرضيًا فقط ، فإن العلاج الكامل غير ممكن. يعتمد الشخص المصاب على العلاج مدى الحياة ، ولهذا السبب فإن خيارات الرعاية اللاحقة محدودة للغاية.
كقاعدة عامة ، بسبب نقص A-N-acetylgalactosaminidase ، غالبًا ما يعتمد المريض على تناول الأدوية والمضادات الحيوية. يجب أخذ التفاعلات المحتملة مع الأدوية الأخرى في الاعتبار ، على الرغم من أنه لا ينبغي تناول المضادات الحيوية مع الكحول. إذا تعرضت لنوبة ، يجب عليك الذهاب إلى المستشفى أو الاتصال بطبيب الطوارئ مباشرة.
علاوة على ذلك ، يجب الحفاظ على الرئتين لتجنب الالتهاب. لذلك ، يجب ألا يدخن المرضى الذين يعانون من نقص A-N-acetylgalactosaminidase تحت أي ظرف من الظروف. إن اتباع نظام غذائي صحي ونمط حياة صحي بشكل عام لهما تأثير إيجابي للغاية على مسار المرض.
إذا كان المريض يرغب في إنجاب الأطفال ، فإن الاستشارة الوراثية مفيدة لمنع انتقال المرض إلى الأطفال. نظرًا لأن المرضى غالبًا ما يعانون من تقييد الحركة ، فإن العلاج الطبيعي مفيد للتخفيف من ذلك. يمكن أيضًا أداء العديد من التمارين في منزلك.
يمكنك أن تفعل ذلك بنفسك
نقص A-N-acetylgalactosaminidase الموجود هو مرض عضال. لذلك فإن العلاج الطبي ضروري. لا يمكن أن تستند إجراءات العلاج الذاتي إلا على الأعراض لجعل الحياة اليومية أسهل. من المهم أن يراقب الوالدان الطفل عن كثب.
يمكن للوالدين مساعدة أطفالهم بشكل أفضل من خلال التفاهم والصبر والحب والرعاية. علاوة على ذلك ، يعتمد العلاج المهني وعلاج النطق دائمًا على مفهوم مزدوج: العلاج في الموقع مع الأخصائي وأداء التمارين في المنزل. يجب على الآباء أن يكونوا نشطين هنا باستمرار وبصبر. يجب ضمان اتباع نظام غذائي غني بالفيتامينات والمعادن وممارسة التمارين الرياضية بانتظام والكثير من الهواء النقي. هذا يقوي جهاز المناعة ويقلل من خطر الإصابة. إذا كان الشخص يميل إلى النوبة ، فلا ينبغي أن يبقى بمفرده لفترة طويلة ويجب فحصه لمعرفة ما إذا كان بإمكانه إيذاء نفسه في حالة حدوث نوبة.
في معظم الأوقات ، لا يستطيع المرضى التعامل مع الحياة اليومية بأنفسهم ويحتاجون إلى رعاية مستمرة. إذا كان الآباء لا يستطيعون تحمل ذلك - خاصة مع الأطفال الأكبر سنًا والبالغين - فلا ينبغي أن يخافوا من طلب المساعدة. يمكن أن يأخذ هذا شكل ممرضة أو التنسيب في منشأة مناسبة. إذا كنت لا تزال ترغب في إنجاب الأطفال ، فيوصى باستشارة أخصائي ، لأن المرض وراثي.
.jpg)


.jpg)

.jpg)




.jpg)


.jpg)